paired end sequencing reads
Now lets get started. To start analysis of paired end Illumina sequence targeted amplicon data you need to create several files describing your data input and the raw sequences files which should be de-multiplexed on the Illumina barcodes already and are in a directory and gzipped.

Methode Sequencage Assemblage Genomique Fonctionnelle Vegetale Enseignement Et Recherche Biochimie Universite Angers Emmanue Biochimie Genomique Enseignement
Today most researchers use the paired-end approach.

. Paired-end reading improves the ability to identify the relative positions of various reads in the genome making it much more effective than single-end reading in resolving structural rearrangements such as. Paired-end 150 means that one read of 150 bases in size is generated from each end of the fragment through the inserted middle piece of target DNA from both directions for a total of 2 paired reads per fragment. Paired-end reading In single-end reading the sequencer reads a fragment from only one end to the other generating the sequence of base pairs.
Pairs come from the ends of the same DNA strand. The Illumina paired-end sequencing technology can generate reads from both ends of target DNA fragments which can subsequently be merged to increase the overall read length. In general paired-end reads tend to be in a FR orientation have relatively small inserts 300 - 500 bp and are particularly useful for the sequencing of fragments that contain short repeat regions.
Paired-end tags exist in PET libraries with the intervening DNA absent that is a PET represents a larger fragment of. T he term paired ends refers to the two. Paired-end sequencing means sequencing both ends of the cDNA fragments and aligning the forward and reverse reads as read pairs Figure 8.
Coli genome is 35 nt and 60 nt for the. Paired-end reads are preferable for de novo transcript discovery or isoforms expression analysis as well as to characterise poorly annotated transcriptomes. Paired-end sequencing facilitates detection of genomic rearrangements and repetitive sequence elements as well as gene fusions and novel transcripts.
Assembling the paired-end reads from each RAD site sequence one RAD sequence at a time resulted in 53296 contigs with an N 50 length of 407 nucleotides Figure 2A Files S1 S2 S3. - Paired end gives an idea of the size of the insert and the diectionality of the mapping to the sequence assembly algorithms. The paired-end reads associated with each RAD site were extracted and the 301000 sequences sent to the de novo assembly program Velvet.
It has very nice and simple illustrations along with explanations on the terminology used in paired-end sequencing. Illumina gets sequence data from both strands of input sequence which means it outputs data from both ends of the input and is normally reported two files R1 and R2 often refereed to as mates files R1first mates R2second mates. An analysis by Whiteford et al.
What does paired end reads mean. Paired-end DNA sequencing reads provide high-quality alignment across DNA regions containing repetitive sequences and produce long contigs for de novo sequencing by filling gaps in the consensus sequence. In conventional paired-end sequencing you simply sequence using the adapter for one end and then once youre done you start over sequencing using the adapter for the other end.
In contrast RNA-seq on short RNAs 200 nt is typically carried out in single-end mode as the additional cost. Due to the way data is reported in these files. The 2 complementary DNA strands are oriented in opposite orientation and sequence reads from either end are generating results of those 2 different strands.
On sequencing using unpaired reads shows that ultra-short reads theoretically allow whole genome re-sequencing and de novo assembly of only small eukaryotic genomes. Paired-end sequencing allows users to sequence both ends of a fragment and generate high-quality alignable sequence data. Since paired-end reads are more likely to align to a reference the quality of the entire data set.
Sequencing kits for HiSeq systems are available with a single-read or paired-end flow cell. This means your two reads are the reverse complement of the 100 3-most bases of the Watson strand and the Crick strand. Ad Gene Expression Profiling Chromosome Counting Epigenetic Changes Molecular Analysis.
The control software performs Read 1 any index reads and then Read 2 based on the parameters provided for the run in the sample sheet or during run setup. For those not familiar with paired-end reads check out this post. With paired-end sequencing after a DNA fragment is read from one end the process starts again in the other direction.
Paired-end DNA sequencing also detects common DNA rearrangements such as insertions deletions and inversions. Paired-end RNA sequencing RNA-seq is usually applied to the quantification of long transcripts such as messenger or long non-coding RNAs in which case overlapping pairs are discarded. For the first test I took some sequence from the human genome hg19 and created two 100 bp reads from this region.
Paired end や mate pair という用語はどのようにライブラリが作られたかどうやってシーケンスされたかを示します. Library preparation protocols -- In short PE protocols attach an adapter SP1 to the fwd end and another adapter SP2 to the reverse end. This raw directory will not be modified in any way.
Illumina Paired End Sequencing. Mate-pair fragments are generally in a RF conformation contain larger inserts 3 kb and enable sequence coverage of genomic regions containing large structural. Chaisson Brinza and Pevzner 2 recently determined that the paired read length threshold for de novo assembly of the E.
In addition to producing twice the number of sequencing reads this method enables more accurate read alignment and detection of structural rearrangements. 例えばゲノム DNA を. In paired-end reading it starts at one read finishes this direction at the specified read length and then starts another round of reading from the opposite end of the fragment.
The differences between PE and MP reads include. There already exist tools for merging these paired-end reads when the target fragments are equally long. These reads are assumed to be identical to the 100 5-most bases.
In paired-end reading it starts at one read finishes this direction at the specified read length and then starts another round of reading from the opposite end of the fragment. This aids in prediction of inversions deletions and. The first sequencing step is started by targeting SP1 to generate the forward read.
Paired-end tags are the short sequences at the 5 and 3 ends of a DNA fragment which are unique enough that they exist together only once in a genome therefore making the sequence of the DNA in between them available upon search or upon further sequencing. Fast and Accurate Next-Generation Sequencing Results Enabled by Ion Torrent Technology. For all other systems sequencing kits include a paired-end flow cell.

Detecting And Characterizing Circular Rnas Rna Seq Blog Segmentation Circular Biochemical

Experimental Designs That Take Advantage Of High Throughput Sequencing To Generate Datasets Include Rna Sequencing Gene Sequencing Rna Sequencing Experiments

Paired End Sequencing Next Generation Sequencing Repeated Reading Sequencing

In This Study Researchers From Stanford University Generated Two Paired End Rna Seq Datasets Of Differing Read Lengths 2 7 Rna Sequencing Long Reads Analysis

Exon Based Strategy To Identify Differentially Expressed Genes In Rna Seq Experiments Experiments Statistical Analysis Gene

Esnv Detect A Computational System To Identify Expressed Single Nucleotide Variants From Transcriptome Sequencing Data Next Generation Sequencing Sequencing
![]()
Pin On Lncrna

Shrna Seq Data Analysis With Edger Rna Seq Blog Analysis Data Analysis Step Function
Researchers At Agroparistech Propose To Use The Pruned Dynamic Programming Algorithm For Seq Experiment Outputs This Requires The Segmentation Algorithm Data

Badge A Novel Bayesian Model For Accurate Abundance Quantification And Differential Analysis Of Rna Seq Data Rna Seq Blog Analysis Data Novels

How Do You Put A Genome Back Together After Sequencing Genome Sequencing Learning Resources

Rcount Simple And Flexible Rna Seq Read Counting Rna Seq Blog Reading Flexibility Gene Expression
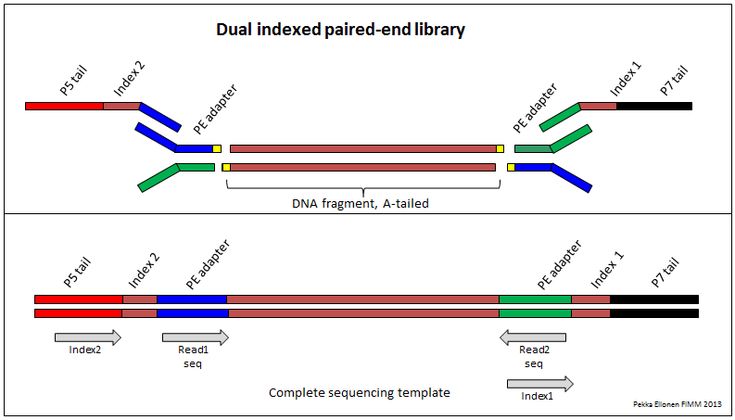
Demultiplexing Dual Indexed Miseq Fastq Files Seqanswers Data Science Index Hypothesis

In The Current Work Researchers From The Institute Of Applied Genomics Italy Evaluate Nine Different Trimming Alg Data Visualization Data Analysis Evaluation

Rna Extraction Method Read Length And Sequencing Layout Single End Versus Paired End Contribute Strongly To Var Interactive Notebooks Method Gene Expression

Pin On Services

Personal Genome Assembly Is A Critical Process When Studying Tumor Genomes And Other Highly Divergent Sequences The Data Visualization Reference Visualisation

Exon Based Strategy To Identify Differentially Expressed Genes In Rna Seq Experiments Experiments Statistical Analysis Gene

Mapping Mutants By Ngs Next Generation Sequencing Short Reads Generation